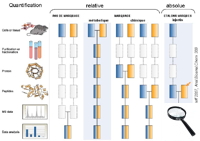

Relative protein quantification consists in measuring the relative differences in abundance for proteins between, most often, two analogous samples in differential conditions,
e.g. "disease"/"healthy" (
DECanBio,
CT-Lymph projects), "wild-type"/"mutant" (
Chloroplast project), "normal environment"/"stressed environment" (
Plantox-Ura project). This can involve large sample series (over two hundred samples), for example to follow variations in protein abundance during a time-course.
Spectral count is a simple, semi-quantitative method. It consists in estimating a protein's relative abundance in a sample based on the number of MS/MS spectra associated with it (more abundant peptides will be fragmented more frequently by the mass spectrometer). This estimation, which is now automated in our process, is included in our identification reports. All other relative quantification methods use intensity measurements, the choice of method will depend on the biological project.
-
Label-free methods: AMT (Accurate Mass Tag) is a label-free method identifying peptides based on their chromatographic retention time and high-precision mass, determined using a Fourier Transform-based mass spectrometer. Quantification does not require peptide fragmentation. This accelerates throughput, which is a definite advantage for projects with large numbers of samples (
AT-Chloro,
DECanBio projects).
-
Labelled methods: these methods consist in using stable isotopes to mark the proteins - or peptides - in one of the samples to be compared (
SILAC, 15N) (
Algomics project). Because of the label, samples can then be mixed (labelled with non-labelled) before mass spectrometry analysis. This reduces variability due to differences introduced during sample preparation, but it also increases the complexity of search algorithms, and
ad hoc computing developments are necessary to treat the corresponding data.