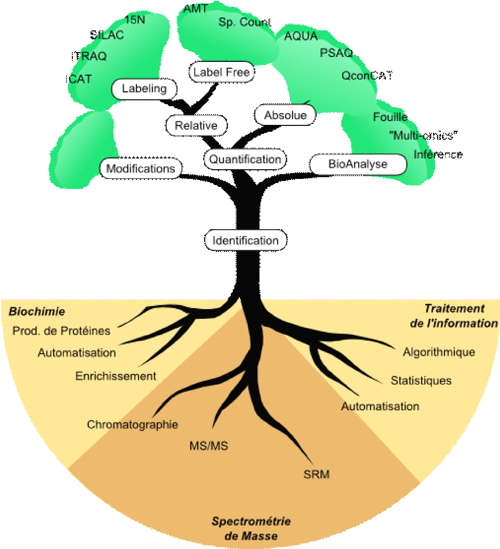
 Mass spectrometry methods
Mass spectrometry methods make it possible to analyse all the proteins present in a selected biological sample. Analysis first involves identifying proteins based on their amino acid sequence. Complementary methods exist to quantify protein expression, analyse post-translational modifications, and detect specific peptides.
Computing power is crucial to treat the data generated by mass spectrometers. The computing team manages the infrastructure handling the volumes of data generated (up to 200 Go/month) and develops software solutions for data treatment.
Our expertise in
bioinformatics allows us to interpret validated identification results with a view to the biological question asked. Inferring biological information requires appropriate bio-analytical methods, particularly for analyses generating data lists relating to several thousand proteins.