 Identification des protéines
Identification des protéines
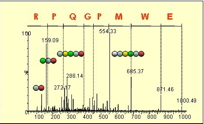
La plupart des méthodes protéomiques actuelles reposent sur la spectrométrie de masse (MS) dédiée à la mesure de la masse moléculaire d'une espèce d'intérêt. La spectrométrie de masse peut également être utilisée pour dériver des informations structurales en fragmentant les molécules analysées afin de déterminer les masses de leurs fragments; on parle alors de spectrométrie de masse en mode tandem (ou MS/MS). Typiquement, une étape de digestion enzymatique des protéines d'intérêt précède l'analyse en masse: une enzyme spécifique comme la trypsine, coupe les protéines en des positions particulières de leur séquence et produit un mélange complexe de peptides (petits fragments de protéines). Afin de gérer la complexité de ces échantillons, les mélanges de peptides sont séparés par chromatographie liquide (LC) avant leur injection dans la source du spectromètre de masse. Pour chaque peptide, les analyses MS et MS/MS permettent d'accéder à la mesure de sa masse et à celle de fragments. Ces informations permettent alors de repérer dans les banques de séquences protéiques la protéine dont est issu le peptide.
Les étapes de préparation de l'échantillon et les analyses au spectromètre sont réalisées automatiquement grâce à la
plate-forme instrumentale.
Quantification relative des protéines
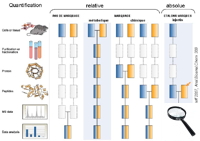
La quantification relative consiste à mesurer les différences relatives d'abondance des protéines entre, le plus souvent, deux échantillons analogues en conditions différencielles, par exemple « malade » / « sain » (projets
Decanbio,
CT-Lymph), « sauvage » / « mutant » (projet
Chloroplastes), « environnement normal » / « environnement de stress » (projet
Plantox-Ura). Une série de plus de deux échantillons peut être considérée, par exemple pour suivre les variations d'abondance des protéines au cours d'une cinétique.
Une méthode de quantification simple, semi-quantitative, est celle du
Spectral Count : elle consiste à estimer l'abondance d'une protéine dans l'échantillon à travers le nombre de spectres MS/MS qui lui sont associés (plus un peptide est abondant, plus il a de chance d'être fragmenté dans le spectromètre). Cette estimation, désormais automatisée, est intégrée dans nos compte-rendus d'identifications. Toutes les autres méthodes font appel à des mesures d'intensité et la méthodologie choisie dépendra du projet biologique.
-
Méthodes sans marquage : la méthode AMT (
Acurate Mass Tag) est une méthode sans marquage qui permet l'identification des peptides, sur la base de leur temps de rétention chromatographique et de leur masse déterminée de façon ultra-précise grâce à l'utilisation d'un spectromètre de masse à transformée de Fourier. La quantification ne nécessite pas de fragmenter les peptides ce qui permet un débit d'analyse élevé, un avantage certain pour les projets comportant de grands nombres d'échantillons (projets
AT-Chloro,
Decanbio)
-
Méthodes avec marquage : ces méthodes consistent à marquer par des istopes lourds les protéines - ou peptides - d'un des deux échantillons à comparer. (
SILAC, 15N) (projet
Algomics). L'intérêt est de pouvoir ensuite mélanger les échantillons (marqués et non marqués) avant l'analyse au spectromètre de masse et par conséquent, de diminuer les variations liées à la préparation de l'échantillon. En revanche, le marquage augmente la complexité des algorithmes de recherche et rend nécessaires les
développements informatiques ad hoc.
Quantification absolue des protéines
Les méthodes de quantification absolue s'appliquent à des protéines préalablement identifiées (biomarqueurs, toxines, hormones) dont on désire déterminer la quantité exacte dans l'échantillon. Ce sont des méthodes de dosage qui impliquent l'injection dans l'échantillon de peptides-étalon (
AQUA) ou de protéines-étalon (
PSAQ™) isotopiquement alourdis. La méthode PSAQ™ (
Protein Standard for Absolute Quantification) développée au laboratoire consiste à utiliser une protéine entière recombinante, biochimiquement identique à la protéine native. La quantification par PSAQ™ hautement spécifique, sensible et précise, s'est avérée être une excellente alternative aux dosages dépendants d'anticorps. La méthode a été brevetée et est proposée par la société start-up
Promise Advanced Proteomics.
Ces méthodes sont utilisées dans les projets d'évaluation/quantification de biomarqueurs (projet
CardioPSAQ™).
Analyses ciblées
… de protéines en particulier.
L'analyse SRM consiste à détecter des protéines d'intérêt, même dans un échantillon complexe. SRM signifie "
Single Reaction Monitoring" et désigne le mode particulier du spectromètre dans lequel il est configuré pour analyser
uniquement les peptides (mode MS) et les fragments (mode MS/MS) caractéristiques de la protéine étudiée. Un soin particulier est apporté au choix des deux à trois peptides représentatifs de la protéine ("protéotypiques") ainsi que pour chacun de ces peptides, des deux-trois fragments représentatifs. Les analyses SRM sont en général associées à la quantification.
… de modifications en particulier.
Les modifications post-traductionnelles (PTMs) des protéines jouent un rôle prépondérant dans tous les processus cellulaires. L'identification des sites modifiés sur les protéines mais également de la dynamique de ces modifications sont devenues des enjeux majeurs et incontournables de la protéomique. Aussi, certains de nos projets sont articulés autour de l'analyse ciblée de PTMs particulières, principalement : les phosphorylations, les acétylations (projet
EpiGam), les méthylations (projet
EpiGam), les glycosylations. Les PTMs peuvent être étudiées dans le cadre d'acquisitions de type LC-MS/MS ou SRM.
… de peptides en particulier.
L'identification des
peptides N-terminaux des protéines permet de reconnaître s'il y a eu maturation de la protéine comme typiquement l'excision d'une préséquence signal associée au transfert de la protéine dans la mitochondrie, ou le réticulum endoplasmique, ou le chloroplaste (chez les organismes photosynthétiques). L'identification de ces peptides n'est pas triviale. Elle est conditionnée à des méthodes particulières, d'enrichissement de l'échantillon en ces peptides et/ou de critères de recherche et de validation adéquats lors de la confrontation avec les banques de séquences. Elle a été appliquée pour constituer un set d'entraînement pour un outil de prédiction de localisation subcellulaire chez les Algues.