Au sein d’un organisme vivant, un type cellulaire se différencie d’un autre par un programme d’expression de gènes qui lui est propre. Les gènes sont codés par l’ADN qui s’enroule autour de protéines nommées histones afin de former une structure appelée la chromatine. L’expression des gènes est étroitement contrôlée par la modification dynamique des histones
via l’ajout de groupes chimiques covalents sur certains acides aminés. Les modifications les plus connues sont la méthylation et l’acétylation des lysines d'histones. Leur diversité s'est largement accrue ces dix dernières années avec la découverte d’une large palette d’acylations, structures assez proches de l’acétylation mais qui confèrent aux lysines modifiées des propriétés physico-chimiques différentes. Ces acylations sont ajoutées sur les lysines à partir des petites molécules (métabolites) Acyl-CoA correspondants. Une question clef qui a émergé de la découverte de ces nouvelles modifications que l’on appelle marques épigénétiques, est de savoir si elles assurent des fonctions redondantes avec l’acétylation ou si elles sont dotées de rôles spécifiques, notamment dans la dynamique de la structure de la chromatine (ouverte ou compactée) et le contrôle de l’expression des gènes.
Parmi les acylations, la crotonylation est une modification des résidus lysine qui consiste en l'ajout d’un groupe crotonyle dont la particularité est sa structure rigide plane. Pour mieux comprendre le rôle de cette marque épigénétique ignorée jusqu’en 2011, des chercheurs de notre laboratoire ont analysé sa dynamique au cours du processus de différentiation cellulaire que constitue la spermatogenèse murine. Par analyse protéomique, ils ont en particulier observé que la crotonylation de la lysine 27 de l’histone H3 (H3K27cr) était d'abondance similaire à l’acétylation de ce même acide aminé (H3K27ac). L’acétylation des lysines d'histones a été explorée depuis plus de 50 ans, et H3K27ac est connue pour être un marqueur de l'expression active des gènes lorsqu'elle est présente en amont de leur séquence. Étant donnée l'abondance notable de H3K27cr, il semblait très opportun de mettre enfin en lumière ses fonctions cellulaires par rapport à celles de H3K27ac. Les chercheurs ont alors obtenu la localisation sur le génome de cette nouvelle marque d'histone et l’ont comparée à celle de H3K27ac, ainsi qu’à celle de protéines impliquées dans la régulation de la transcription des gènes. Ils ont observé que ces deux marques fonctionnaient souvent en synergie, induisant une expression génique maximale. Cependant, certaines régions du génome portent davantage l’une ou l’autre marque, ce qui a pour effet d’attirer préférentiellement des types différents de régulateurs de la transcription.
Cette analyse intégrée des données de type « omique » fournit un niveau de compréhension de la régulation de l'expression génique de ces deux marques jamais atteint à ce jour, et révèle les actions synergiques et spécifiques de chaque modification d'histone. Connaître l’ensemble des formes modifiées, ainsi que les protéines qui s’y lient, est essentiel à la compréhension des mécanismes moléculaires régulant la transcription des gènes à l’œuvre dans des processus de développement normaux, mais aussi dans de nombreuses pathologies.
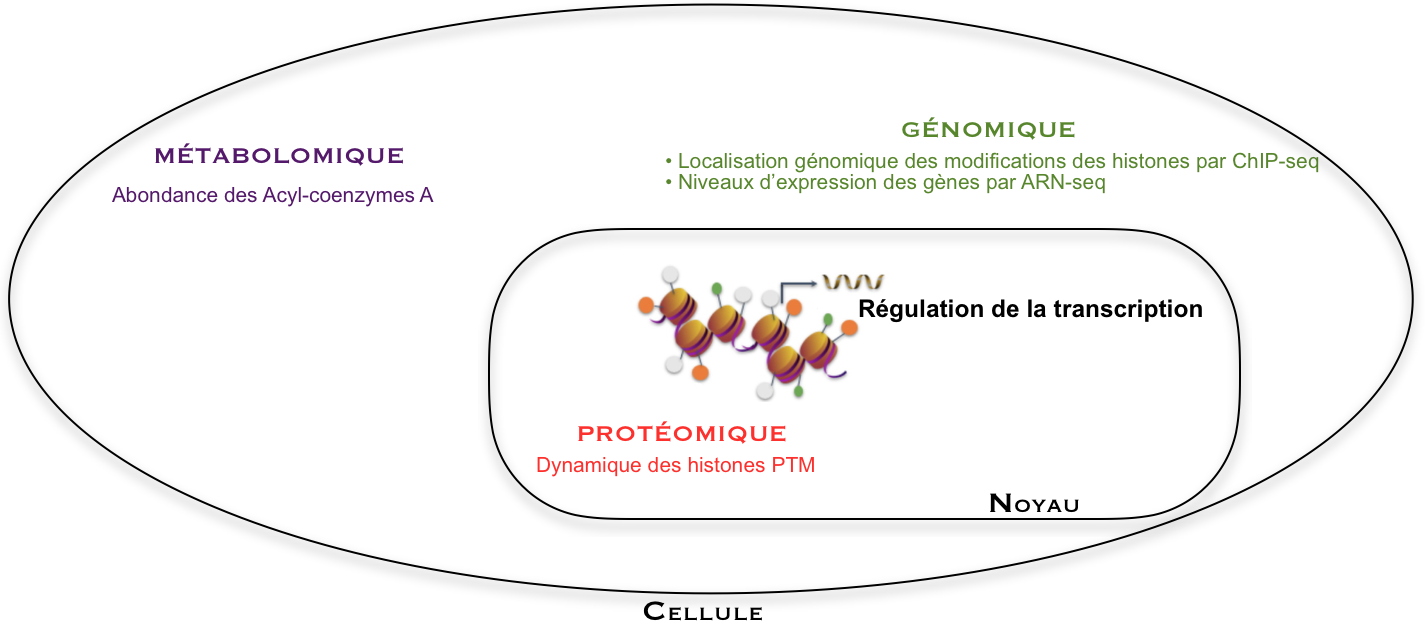
L’expression des gènes est analysée par analyse génomique de type RNA-seq (une technique de séquençage). Elle est régulée par des modifications d’histones, en particulier sur des lysines dont la position sur la séquence des histones et le niveau de modification peuvent être appréhendés par analyse protéomique. Les acylations de lysines sont ajoutées à partir des cofacteurs Acyl-CoA, dont les abondances sont quantifiées par analyse métabolomique. La position d’une marque d’histone sur le génome (comme H3K27cr), est obtenue par un autre type d’analyse génomique (ChIP-seq, une technique permettant d’étudier les interactions ADN/protéine à l’échelle du génome). Ces diverses analyses ont été combinées dans ce travail pour mieux comprendre les mécanismes de régulation de l’expression de gènes.