Ces dernières années, avec l’installation d’une plate-forme instrumentale de très haute technicité et la création d’une équipe dédiée, ll'équipe EDyP a réalisé une des premières implémentations européennes d’une technologie d’analyse protéomique à haut débit, appelée « AMT tags », avec l’objectif de contribuer à améliorer nos connaissances fondamentales en biologie cellulaire et de découvrir de nouveaux biomarqueurs précoces de pathologies.
La Transcriptomique et la Protéomique dénomment l’ensemble des méthodes permettant d’étudier à large échelle les ARNm et les protéines, respectivement. Alors que les méthodes d’analyse transcriptomique ont atteint une robustesse et un débit élevés, la protéomique demeure un domaine en évolution permanente. En effet, la diversité chimique des protéines, associée à l’ensemble des modifications dont elles peuvent être l’objet (clivage, oxydation, méthylation, phosphorylation, etc), représentent un défi technologique considérable pour les chercheurs. Ainsi, d’après le «
Human Proteome Initiative », les quelques 21 000 gènes humains encodent environ un million de formes protéiques différentes! A cette diversité de composés présents dans un échantillon biologique donné, il faut ajouter leur large gamme d’abondance. Ainsi, par exemple, l’albumine dans le plasma humain est-elle 1010 fois plus abondante que les protéines les moins représentées. Mesurer une telle diversité pourrait être comparé à tenter de peser un éléphant et un moustique sur la même balance!
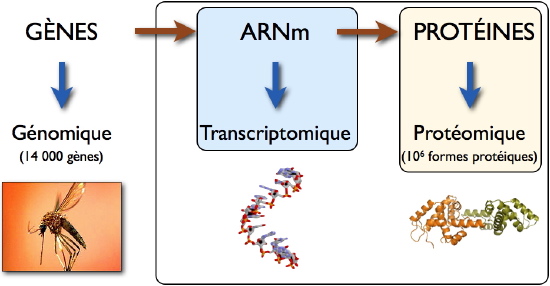
Figure : La protéomique est définie comme étant l'étude systématique de l'ensemble des protéines présentes dans un échantillon biologique dans une situation physiologique ou pathologique donnée.
La plupart des méthodes protéomiques actuelles reposent sur la spectrométrie de masse (MS) conçue pour mesurer la masse moléculaire d’une espèce (ou d’un mélange d’espèces) d’intérêt. La spectrométrie de masse peut également être utilisée pour dériver des informations structurales en fragmentant les molécules analysées afin de déterminer les masses de leurs fragments ; on parle alors de spectrométrie de masse en mode tandem (ou MS/MS). La spectrométrie de masse est typiquement utilisée en protéomique après une étape de digestion enzymatique des protéines d’intérêt : une enzyme spécifique (la trypsine) coupe les protéines en des positions particulières de leur séquence (après les acides aminés basiques), et produit un mélange complexe de peptides (petits fragments de protéines). Afin de gérer la complexité de ces échantillons, les mélanges de peptides sont séparés par chromatographie liquide (LC) avant leur injection dans la source du spectromètre de masse. Pour chaque peptide, les analyses MS et MS/MS permettent d’accéder à la mesure de sa masse et à celle de fragments. Ces informations permettent alors de repérer dans les banques de données protéiques (Uniprot) la protéine dont est issu le peptide. Cette approche conventionnelle, connue sous le nom de « shotgun », souffre de limitations intrinsèques : efficacité, fiabilité des données, sous-échantillonnage des mélanges complexes, sur représentation de certains peptides par rapport à d’autres.
Pour surmonter ces problèmes, une stratégie innovante a été développée qui permet l’identification de peptides sur la base de leur temps de rétention chromatographique et de leur masse déterminée de façon ultra-précise grâce à l’utilisation d’un spectromètre de masse à transformée de Fourier. Cette méthode, appelée «
Accurate
Mass and
Time tags », a récemment démontré un fort potentiel en recherche protéomique, en particulier un débit d’analyse élevé, ce qui rend possible la gestion d’analyses protéomiques quantitatives sur de grands nombres d’échantillons.
Grâce à l’environnement qui existe au sein de l'équipe EDyP, notre groupe a été en mesure d’implémenter et de tester la méthode AMT au travers d’une étude de l’enveloppe du chloroplaste d’Arabidopsis thaliana, une plante utilisée comme modèle en biologie végétale. En collaboration avec le laboratoire Physiologie Cellulaire & Végétale de notre institut, nous avons obtenu la première base de données des protéines du chloroplaste et de ses sous-compartiments (plus de 1 300 protéines). Couplées à des méthodes de fractionnement biochimique des échantillons réalisées par l’équipe de Norbert Rolland (laboratoire Physiologie Cellulaire & Végétale), des informations de localisation subcellulaire ont été obtenues et utilisées afin de rechercher la fonction de plusieurs de ces protéines chloroplastiques par analyse protéomiques comparative de divers mutants d’Arabidopsis thaliana.
Nous avons également utilisé la méthode AMT pour établir les profils différentiels d’abondance protéique entre des cholangiocarcinomes (tumeur des voies biliaires) micro-disséqués par laser et des cellules saines (Collaboration avec l’équipe Inserm U785 (C. Bréchot, F. Demaugre, V. Thiers, A. Dos Santos). Plusieurs biomarqueurs potentiels de cholangiocarcinome ont ainsi été mis en évidence. Finalement, en permettant d’analyser des échantillons issus de cohortes de patients, cette méthode est amenée à jouer un rôle majeur en recherche clinique. Nous l’utilisons actuellement dans le cadre d’un programme de recherche européen (DECanBio, 7ème PCRD), coordonné par l'équipe, dont l’objectif est de découvrir des biomarqueurs protéiques du cancer de la vessie.