Le séquençage du génome d’un organisme doit être suivi de son annotation syntaxique (repérage des gènes et définition de leur structure) et fonctionnelle (analyse des fonctions des produits des gènes). Une partie de l’activité du laboratoire EDyP est tournée vers l’utilisation de la protéomique comme outil d’annotation des génomes.
Un protéome est défini comme l’ensemble des protéines codées par un génome dans des conditions données (carence, stress, état physiologique (cancer), etc). La protéomique a pour vocation d’approfondir la compréhension des mécanismes biologiques en se consacrant à l’étude des protéomes. Une analyse « classique » en protéomique repose sur la confrontation de données expérimentales issues des spectres de masse avec les répertoires de séquences protéiques déjà connues ou prédites. Dans le cas où les protéines seraient mal décrites ou non représentées dans ces répertoires, il devient pertinent d’exploiter les données issues des spectromètres de masse en explorant directement la séquence du génome, et non les séquences protéiques répertoriées. Cette stratégie permet, au-delà du processus d’annotation syntaxique du génome, d’identifier avec justesse et de façon automatisée un plus grand nombre de protéines et de décrypter des mécanismes moléculaires nouveaux mis en jeu dans le monde du Vivant.
Au-delà de la possibilité par laquelle les données résultant des analyses protéomiques permettent de sonder les banques protéiques, l'équipe EDyP de notre laboratoire (Étude de la Dynamique des Protéomes) a développé en collaboration avec l’Inria (action
Helix) et la société
Cogenics un « pipe-line » informatique (PepLine)
[1] qui permet de confronter directement les données protéiques aux séquences génomiques brutes.
Dans une analyse protéomique, l’échantillon protéique est digéré par une protéase spécifique, la trypsine, et les peptides obtenus subissent dans le spectromètre de masse une fragmentation privilégiée au niveau de la liaison peptidique. Ainsi, chaque spectre généré contient l’information de séquence d’un peptide fragmenté (spectre MS/MS,
Figure 1). Cependant, la complexité des spectres rend la lecture des peptides difficile. La solution retenue
[2] par EDyP consiste à lire partiellement le spectre et à associer à la courte séquence en acide aminés lue les masses adjacentes en
N- et
C-terminal. Ce triptyque, appelé PST (Peptide Sequence Tag), reconstitue une représentation du peptide entier.
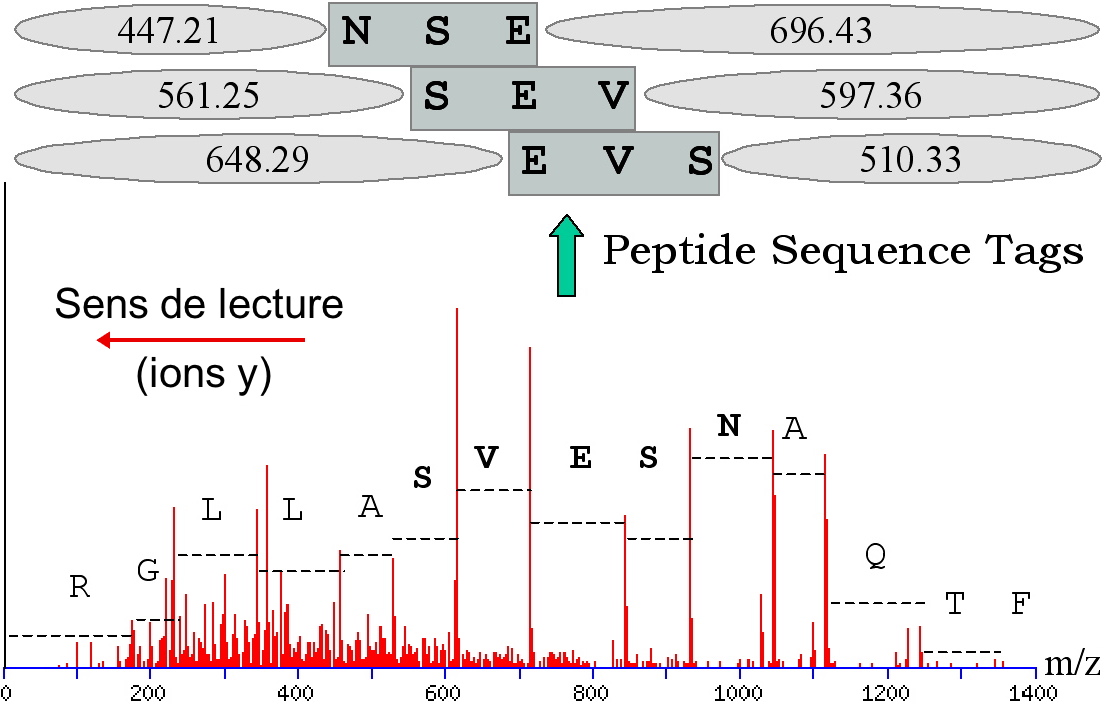
Figure 1 : Spectre MS/MS issu de la fragmentation d’un peptide. Ce type de spectre permet d’accéder à sa séquence en acides aminés par une lecture en « barreaux d’échelle » où la différence entre deux pics majoritaires correspond à la masse d’un résidu d’acide aminé. Les PST sont le résultat d’une lecture partielle de ces spectres, dans lesquels seule une portion de la séquence est déterminée,. On associe à cette séquence deux masses adjacentes, de séquences inconnues, correspondant aux masses N et C terminales du peptide trypsique.
La suite logicielle PepLine est constituée de trois modules dont le premier, Taggor (Figure 2A), a pour tâche de générer plusieurs PST pour chaque spectre. Le second module, PMMatch (Figure 2B), aligne les PSTs sur les séquences du génome. Le troisième, PMClust (Figure 2C) regroupe les positions trouvées (des « hits ») en « clusters » selon une méthode de voisinage. C’est cette étape d’agrégation statistique sur le génome qui confère aux « clusters » une forte probabilité de représenter des zones codantes du génome. De plus, PepLine a été conçu de manière à être capable de détecter les frontières
intron-exon* d’un gène et est donc adapté aux organismes eucaryotes. Il permet de corriger des séquences génomiques et d’identifier de nouveaux gènes.
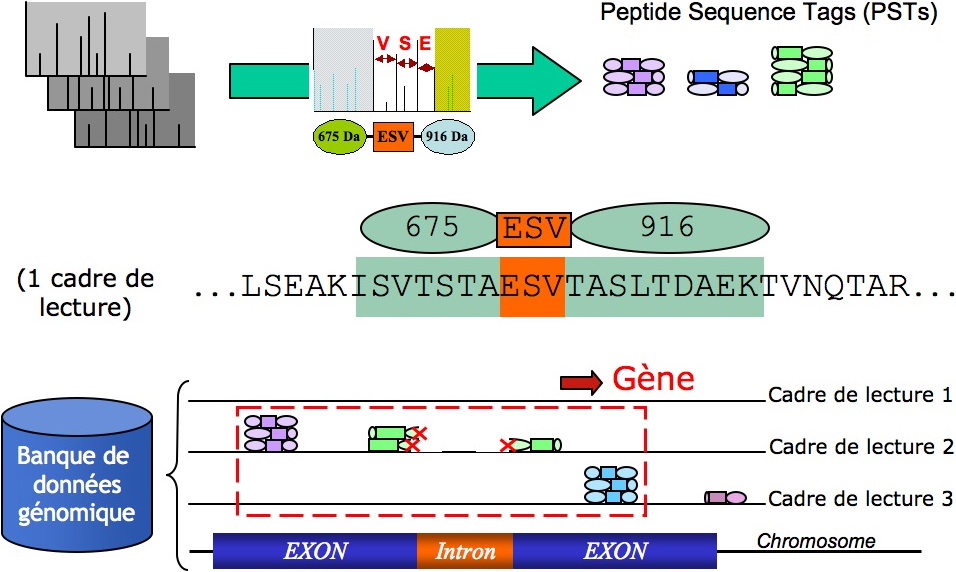
Figure 2 : les trois modules de PepLine :
A - Taggor - Plusieurs PST sont générés par spectre. La partie lexicale de ces PST est restreinte à 3 acides aminés.
B - PMMatch - Un « hit » est complet lorsque la partie lexicale et les deux masses du PSTs s’alignent sur le génome. Des « hits » partiels peuvent être générés si l’une des deux masses ne s’aligne pas sur le génome permettant de révéler les frontières intron-exon.
C - PMClust - Le regroupement des hits en clusters se fait soit sur une seule phase ou bien sur les trois phases d’un même brin selon qu’il s’agit du génome d’un organisme prorcaryote ou eucaryote.
EDyP a fait la preuve de concept de la performance de PepLine.
Cette stratégie est actuellement testée à grande échelle sur le génome de l’algue verte unicellulaire
Chlamydomonas reinhardtii, à la fois modèle et organisme d’intérêt dans le domaine de la production de biocarburants (lipides et hydrogène).
Intron-exon : De manière générale, les gènes sont constitués d'une suite d'exons et d'introns alternés. Chez les organismes eucaryotes, les exons sont les parties des gènes qui codent des protéines.